1. Description of the methods¶
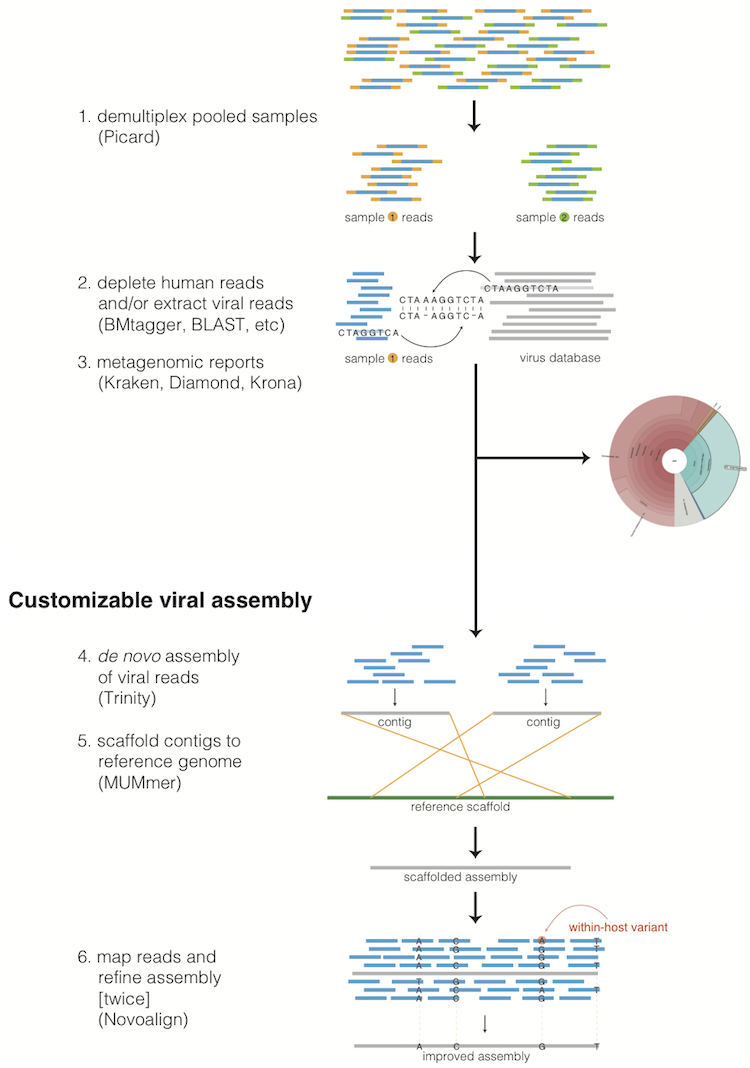
1.1. Taxonomic read filtration¶
1.1.1. Human, contaminant, and duplicate read removal¶
The assembly pipeline begins by depleting paired-end reads from each sample of human and other contaminants using BMTAGGER and BLASTN, and removing PCR duplicates using M-Vicuna (a custom version of Vicuna).
1.1.2. Taxonomic selection¶
Reads are then filtered to to a genus-level database using LASTAL, quality-trimmed with Trimmomatic, and further deduplicated with PRINSEQ.
1.3. Taxonomic read identification¶
Metagenomic classifiers include Kraken and Diamond. In each case, results are visualized with Krona.